Software packages
| Click image to access repository | Software |
|---|---|
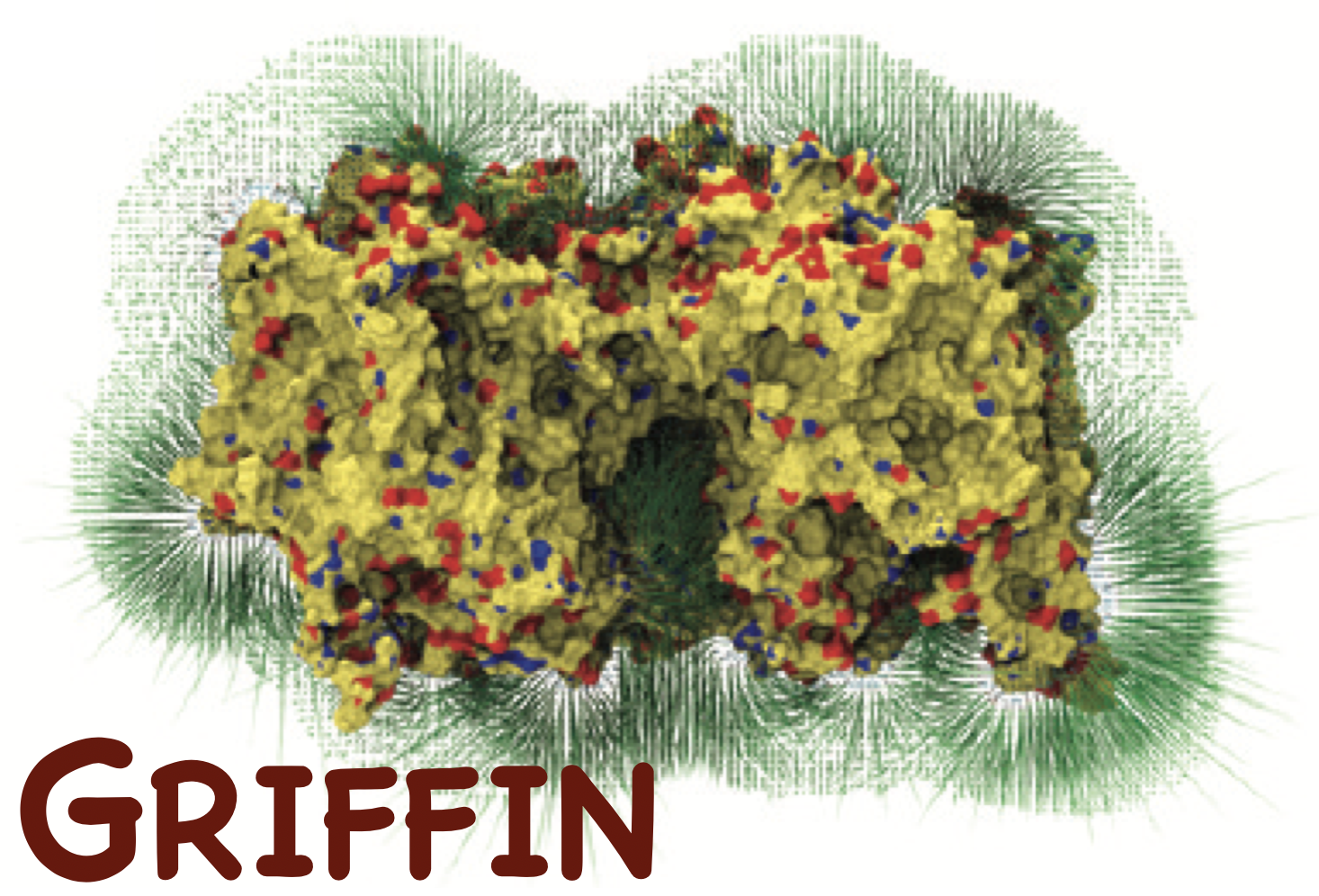 |
Griffin - grid-based implicit force field
Griffin precalculates a potential force field for the molecules that the user selects for implicit treatment.
Implicit means that these molecules are held in a fixed conformation,
while the explicit molecules can move freely in the implicit force field.
Griffin maps Coulomb and van der Waals interactions as well as surface forces, an efficient way for resolving clashes, onto a grid.
For each grid point, Griffin calculates the force experienced by a dummy atom at that position from the entire implicit molecular system.
In its initial version, Griffin is linked to a molecular dynamics simulation of the explicit molecules.
Benefit of this approach is the abillty to resolve clashes even in highly complex topologies.
In addition, the implicit force field approach implies a significant speedup.
|
 |
SmoothT– constructing low energy pathways from simulations SmoothT is a a software for the construction and visualization of transitional pathways between conformational states. The input is an ensemble of molecular conformations in PDB format, along with a list of energy values associated with the conformations. Energies may be physical, statistical or heuristic estimates and combinations thereof. In addition, the user must specify starting and ending conformation, as well as a threshold for the chosen similarity measure, which can be either RMSD or a distance matrix score that is more sensitive to local similarities. |
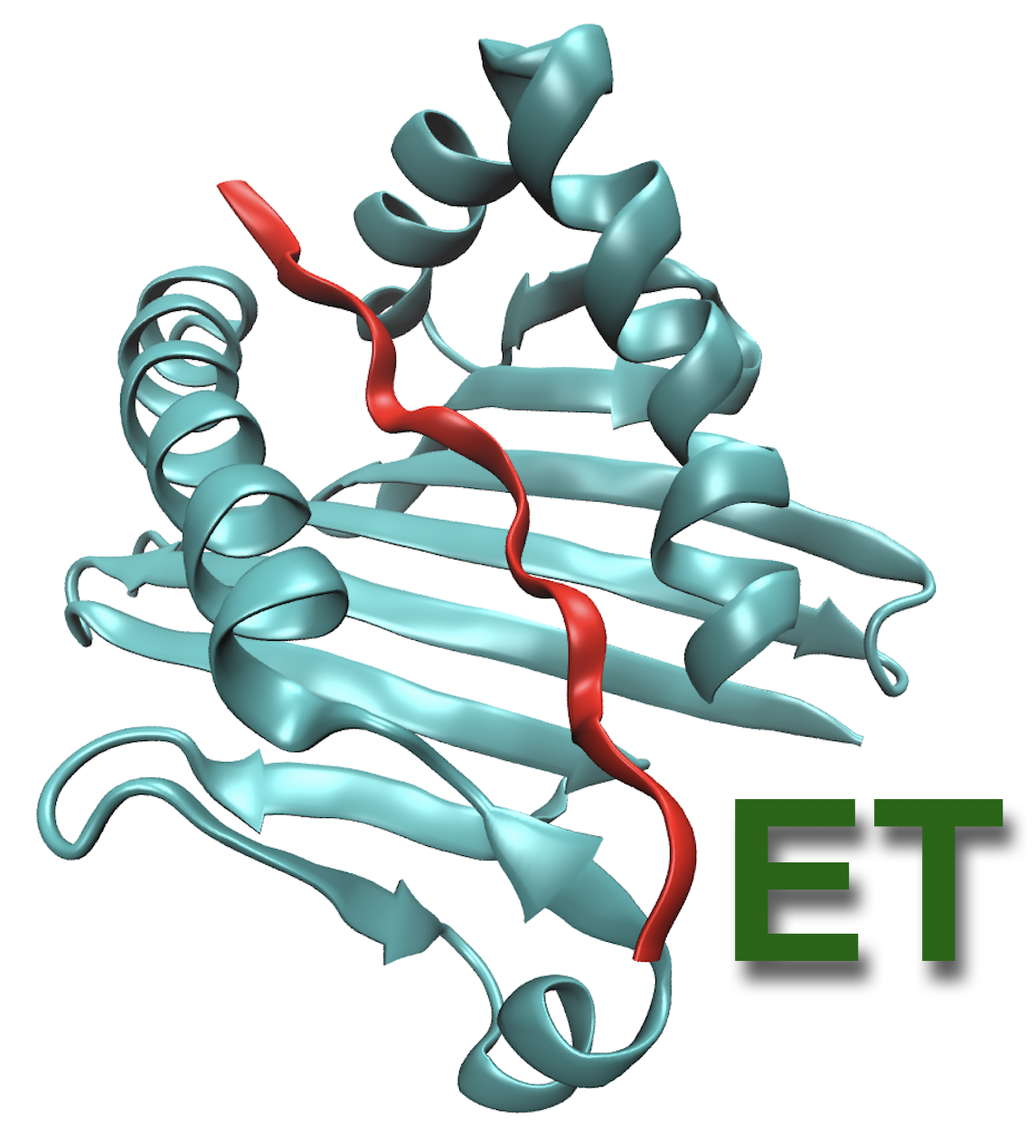 |
EpitopeThreader
The initial activation step of T-cell based immune response pathways of the adaptive immune system is the binding of the T-cell receptor (TCR) to the epitope-presenting major histocompatibility complex (MHC).
Differences in MHC between individuals are essential for the species survival, underscoring the importance of this branch of the immune system.
Since TCR-epitope-MHC structures have a high degree of conservation,
threading is a rapid and accurate method to determine potential activation of the adaptive immune system.
While MHC profiling is a well established standard, sequencing of the TCR repertoire has also been introduced in recent years.
This will allow to predict whether an epitope is a candidate for a vaccine.
|
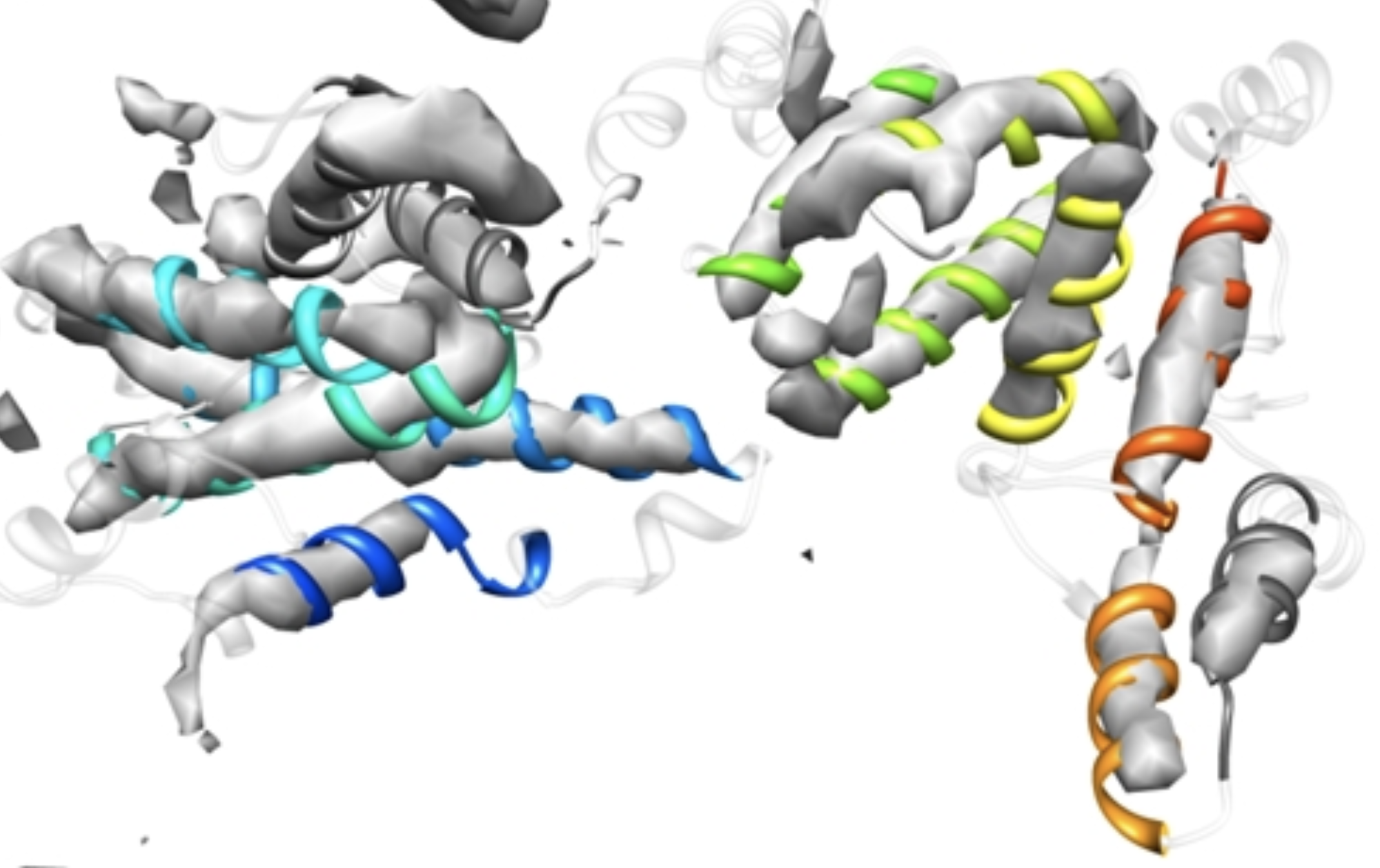 |
bcl::EM-fold
Cryo electron microscopy (EM) has made tremendous progress in the last decade.
Images can be made of very large systems, even entire viruses, sometimes at quite high resolution.
Much more often, however, medium resolutions are obtained.
Resolutions that do not allow to derive a model in atomistic detail directly.
|
| |
AlignmeAlignMe generalizes the concept of similarity by considering evolutionary and biochemical properties. Furthermore, expert knowledge and experimental data can be included as anchors. |
 |
Protein PromptProteinPrompt predicts protein-protein interractions (PPIs) from sequence. We provide two implementations, one using Random Forests, the other Graph Neural Networks. |